

Translate this page into:
Human Gaze Holding and Its Disorders
Address for correspondence Aasef G. Shaikh, MD, PhD, Department of Neurology, Case Western Reserve University. 11100 Euclid Avenue, Cleveland, OH 44110, United States (e-mail: aasefshaikh@gmail.com).
-
Received: ,
Accepted: ,
Abstract
Stabilizing the retinal fovea on the object of interest is the most critical requirement for clear vision. Our brain implements sophisticated neural mechanisms to ensure stable gaze. In this article, I will review contemporary research delineating neural mechanisms for gaze holding. I will then describe various disorders of gaze holding in the context of basic neurophysiology, electrophysiology, and membrane biology. Particular focus is on the basic and translational neuroscience of central nystagmus of ocular motor and vestibular etiologies.
Keywords
neural integrator
nystagmus
cerebellum
brainstem
vertigo
oscillopsia

Introduction
Gaze stabilization on the object of interest is a fundamental requirement for clear vision. The brain has several mechanisms to achieve gaze stabilization. Vestibulo-ocular reflex (VOR) keeps the eyes on the target by shifting them with equal amplitude and velocity as head movements, but in the opposite direction.1 Cerebellar and brainstem-mediated gaze holding mechanisms keep the eyes steady when the head is not in motion. Abnormal spontaneous eye movements such as nystagmus are threats to steady fixation and clear vision. In this article, I will review the fundamental aspects of the physiology of gaze holding. Then I will implement these principles, discussing the pathophysiology of gaze-holding deficits such as nystagmus.
Physiology of Gaze Holding: The Neural Integrator
Scanning of the visual field requires rapid shifts of gaze from one object of interest to the other. Saccadic eye movements rapidly shift the gaze. Once the gaze is directed at the desired scene, it is necessary to hold it steady at the new location so that fovea and brain can capture necessary visual information. Gaze-holding function is guaranteed by a sophisticated neural mechanism which involves the network of neurons that has an ability to perform calculus–mathematical integration. This network, called the neural integrator, in the literal sense, integrates the pulse of neural discharge that corresponds to the eye velocity during saccade into steady-state firing corresponding to the eye position signal during gaze holding. There are two sets of integrators that accomplish such function. When the head is steady, the saccade is integrated by the ocular motor neural integrator residing in prepositus hypoglossi and medial vestibular nucleus for horizontal gaze and interstitial nucleus of Cajal for vertical and torsional eye position.2,3 The second set of integrators, the vestibular integrators, are further upstream from the ocular motor integrator. The vestibular integrator increases the fidelity of the VOR to keep the fovea stable when the head is in motion. The vestibular integrator enhances the faithfulness of the VOR by increasing the bandwidth of frequencies of head oscillations over which the VOR is compensatory. Thus, combined vestibular and eye movement integrators constantly guarantee steady gaze holding whether we are stationary or in motion.
The backbone of neural integrator concept, the conversion of pulse of neural discharge into persistent neuronal activity to form short-term memory, was first proposed in early 18th century by David Hearley and was further elaborated by Alexander Bain in 1800s. Later on, it was proposed that conversion of the pulse of neural discharge to steady-state neural firing requires sustained synaptic activity generated by reverberation in the network of neurons.4,5 This mathematical concept was systematically and thoroughly described in ocular motor and vestibular system by David Robinson in 1970s and 1980s.2,6,7 Contemporary studies concluded that at cellular level the auto feedback between the network neurons and resultant reverberation in the circuit is fundamental to ensure the efficacy of the neural integrator. Such reverberations would lead to gaze position-dependent alteration in the inter-spike membrane potential in the neurons comprising the neural integrator.8-12 For example, the schematic in ►Fig. 1 depicts ideal neural integrator network. The neurons within each network are mutually excitatory. The output of the network to its contralateral counterpart is inhibitory. Mutually excitatory connection within each neural network facilitates conversion of pulse of signal to steady-state neural discharge, while mutual inhibition between the networks on two sides facilitates the coordination of yoked movements of agonist and antagonistic muscle pairs.11 To ensure perfect neural integration, the synaptic strengths of all network neurons must be optimally tuned. Unfortunately, in most biological systems, perhaps due to constant wear and tear, the synaptic strengths cannot be optimally tuned to guarantee perfect output. As a result, the neural integrators become inherently defective or “leaky.” The output of “leaky” integrator is characterized by slow drifts. Fortunately, the drifts are compensated by the external feedback loops around the neural integrator. The cerebellar projections serve as one of the sources of the feedback to the integrator.7,13-15

- Schematic diagram depicting neural network and its reciprocal connections for the function of ideal neural integrator.
Gaze-Evoked Nystagmus: Abnormal Function of Ocular Motor Neural Integrator
A deficit in cerebellar feedback to the ocular motor neural integrator reveals its inherently imperfect behavior causing drifts in the eye position. The drifts are followed by corrective saccades that rapidly realign the eyes toward the target of interest. The drift velocity increases as the eyes move away from the center. The phenomenology of alternating drifts and corrective movements, and their gaze position dependence defines well-characterized gaze-evoked nystagmus.13-15
►Fig. 2 (A-D) depicts an example of gaze-evoked nystagmus. In this figure, the black traces depict eye position plotted on y-axis and corresponding time on x-axis. Each panel illustrates the eye position to aim gaze on the visual target. The eyes are relatively stable in straight-ahead position (►Fig. 2B). However, when the eyes turn 30° to the right, slow leftward drifts deviate them away from the intended target (►Fig. 2A). These drifts are followed by rapid corrective movement in the opposite direction, the quick phase, bringing the eyes back to the target. In contrast, rightward drifts appear when the eyes turn 30° to the left (►Fig. 2C). These drifts are followed by quick phases brining the eyes back to the target. The slow-phase velocity of the drifts (plotted on y-axis ►Fig. 2D) systematically changes with the eye-in-orbit position (plotted on x-axis in ►Fig. 2D). The slow-phase velocity reduces as the eye in the orbit position approaches straight ahead position. All these features are described by suboptimal neural integration of velocity commands and inability to compensate for the imperfection due to defect in the feedback system.14,15
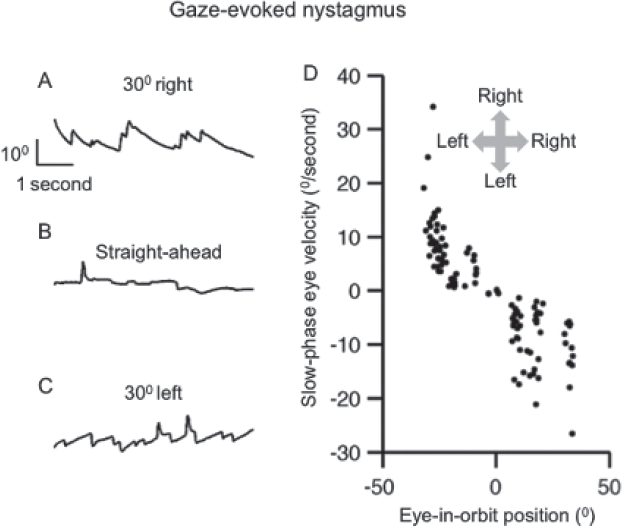
- (A-C) Example of gaze-evoked nystagmus in a patient with cerebellar atrophy. Panel A depicts left drifts and right beats during rightward gaze holding, panel B is straight ahead (stable) gaze holding, while panel C is leftward gaze holding leading to right drifts and left beats. In panels A to C, eye position is plotted on the y-axis while corresponding time is plotted on the x-axis. Only horizontal eye positions are shown. (D) The summary of slow-phase velocity is plotted on the y-axis while corresponding eye-in-orbit position is on the x-axis. Each dot depicts one slow phase. Arrows in inset depict the direction. This figure was reproduced from Shaikh et al.53, with permission.
Vestibular Neural Integrator and the Velocity Storage
The neural integrator, more specific to the vestibular system, called the velocity storage resides further upstream from the ocular motor integrator. The velocity storage, by extending the bandwidth of the angular VOR, enhances the VOR responses at the low-frequency head rotation. Rotational testing measuring the kinematics of per- and postrotational nystagmus is the well-known test of the velocity storage. During constant velocity whole-body rotations (i.e., head rotations), it is expected that the eyes drift in the direction opposite to that of body rotations. However, due to the elastic properties of cupula, the angular velocity signal provided to the brain rapidly decays with ~4-second time constant limiting the brain's ability to persistently perceive ongoing rotations. The velocity storage prolongs the decay of compensatory eye movements beyond that of activity of the vestibular afferents. The velocity storage is not perfect, but its output decays exponentially.16 The time constant of this decay, the velocity-storage time constant, is affected by γ-aminobutyric acid (GABA)-mediated inhibitory projections from the cerebellar cortex (nodulus and ventral uvula).16-22 The GABAb receptor agonist, baclofen, or 4-aminopyridine reduce the velocity-storage time constant in human subjects, while lesions of the nodulus and ventral uvula prolong it.17,19,23 ►Fig. 3 depicts an experiment where velocity-storage function and the influence of GABAB activity modulation with 4-aminopyridine was tested in a healthy human subject. The subject was seated on a three-dimensional servo motor controlled rotating chair (Acutronic, Jona, Switzerland). The head was restrained by custom molded thermoplastic mask (Sinmed BV, Reeuwijk, The Netherlands). ►Fig. 3A depicts results of rotations along earth's vertical axis when the subject was upright. In this instance, the angular vestibular response was primarily through stimulation of the lateral semicircular canals (yaw rotation, schematic inset in ►Fig. 3A). Simultaneously recorded eye movements revealed a rapid rise in slow-phase velocity of the horizontal VOR followed by the instantaneous exponential decline at the time constant of 10 seconds (filled symbols, ►Fig. 3A). The same experiment when repeated 30 minutes after the subject took 10 mg 4-aminopyridine resulted in much faster decay of slow-phase velocity with a time constant of 6 seconds. The results confirmed the presence of strong vestibular velocity storage for horizontal VOR in humans, the results also suggested that horizontal vestibular velocity storage is under cerebellar influence mediated by GABAB receptors.19,24
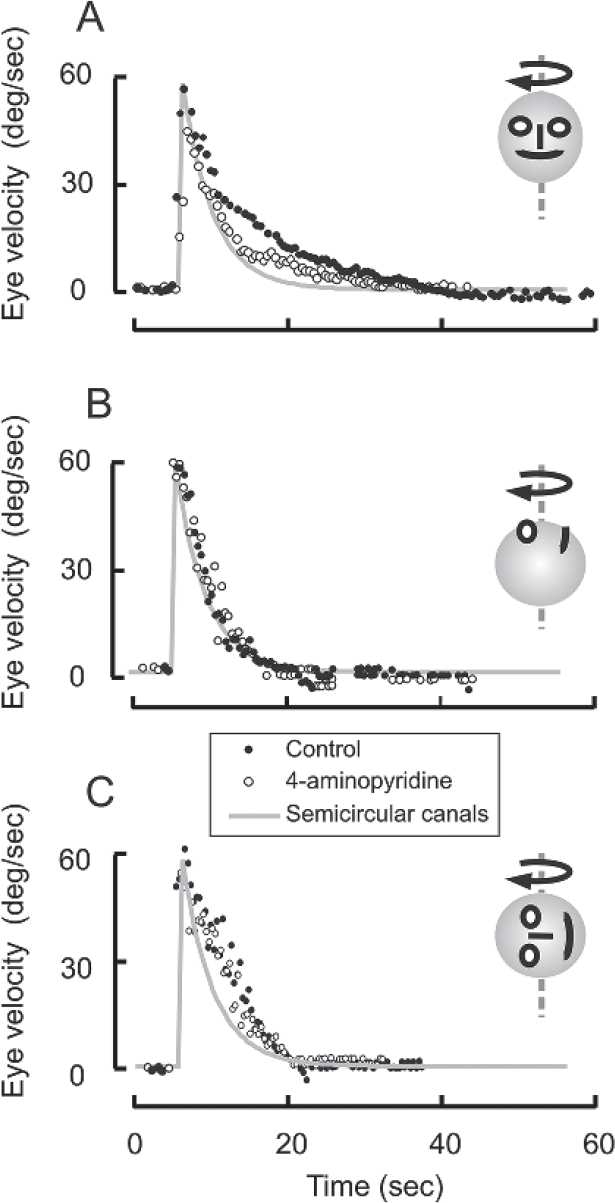
- Effects of constant velocity sustained rotation in yaw plane (A), roll plane (B), and pitch plane (C). Each dot depicts eye velocity (A: horizontal eye velocity, B: torsional eye velocity, C: vertical eye velocity) plotted on the y-axis while corresponding time on the x-axis. Gray line is cupular decay time constant. Filled symbols are eye velocity of rotational (and postrotational) nystagmus before and open symbols after oral intake of 4-aminopyridine. This figure was reproduced from Shaikh, et al.24 with permission.
Such strong GABAergic influence on the velocity storage does not exist in earth's vertical axis while supine (roll rotation, inset ►Fig. 3B) or on the side with left ear facing the ground (pitch rotation, inset ►Fig. 3C). In these conditions when vertical canals are primarily modulated, the VOR was present in torsional (supine rotations) or vertical (ear facing the ground) rotational direction. As depicted in ►Fig. 3B, C, there is no robust change in torsional or vertical VOR suggesting the lack of GABAergic influence.
Gravity-Dependent Nystagmus
Lesions affecting posterior cerebellar vermis, often due to the stroke in the vascular distribution of the posterior inferior cerebellar artery, can cause nystagmus that changes direction with alterations in the head position with respect to the direction of the gravitational force.25-30 The gravity dependence of nystagmus has also been described in degenerative cerebellar syndromes.31-33
The cerebellum calibrates the direction and the amplitude of the vestibular reflexes.31,34,35 Any impairment in such calibration process can lead to gravity-dependent modulation of nystagmus. A two-step process for pathophysiology of gravity-dependent nystagmus was reported.27 It was suggested that disinhibition of the fastigial and vestibular nuclei, after the lesion of posterior cerebellar vermis, leads to their hyperexcitability. Lesion of flocculus, for example, causes gaze-evoked nystagmus due to disinhibition of the neural integrators in midbrain vestibular nuclei.14 The gaze-evoked nystagmus also occurs when there is a breach in the inflow to the cerebellum (from brainstem integrators) that passes through the inferior cerebellar peduncle, in other words there is a disruption of cerebellar feedback pathway around the neural integrator (as noted in the section above). The second step is the gravity-dependent modulation of nystagmus due to impaired central estimate of gravity. It is known that the lesions of the cerebellar nodulus in monkeys impair the ability to align the axis of eye rotation with the gravito-inertial axis during VOR.28-30 Therefore, it was predicted that, in humans too, the lesions affecting the lobule VIII and IX might disrupt the calibration of the axis of eye rotation with respect to gravity. Hence, disinhibition first causes nystagmus, when the axis of eye rotation is not systematically aligned with the earth's fixed axis due to impaired central estimate of gravito-inertial axis that causes gravity-dependent modulation of the nystagmus. Occasionally, the gravity-dependent nystagmus presents with partial features of apogeotropic positional nystagmus, i.e., the quick phase brings the eyes away from the direction of the gravity. ►Fig. 4 depicts an example of gravity-dependent nystagmus in a patient with acute ischemic stroke affecting the cerebellar nodulus, medial cerebellar hemisphere, and lateral medulla, including the inferior cerebellar peduncle (MRI [magnetic resonance imaging] scan in ►Fig. 4). The nystagmus in this patient modulated according to the orientation of the head with respect to gravity. The nystagmus slow-phase velocity was least in left ear-down orientation, but was maximum in right ear-down position. In either ear-down orientation, there was no vertical nystagmus. The direction of nystagmus changed from left beating during left ear-down position to right beating in right ear-down position. In contrast, the slow-phase velocity was comparable upright and supine orientations, in both instances the patient had equal intensity of upbeat and right-beat nystagmus (►Fig. 4).

- Example of gravity-dependent nystagmus during various head orientations with respect to the gravity. Magnetic resonance imaging on the right side of the panel depicts infarct involving lateral medulla and posterior vermis and para-vermis hemisphere.
Although in some occasions the central gravity-dependent nystagmus can resemble common benign paroxysmal positional vertigo (BPPV), they both have key differences. Unlike BPPV, the central positional nystagmus does not follow a peculiar trend in changing its direction with response to head reorientation. Furthermore, the nystagmus related to BPPV appears after latency of seconds, and it habituates within a matter of seconds (i.e., crescendo–decrescendo).1,36
Periodic Alternating Nystagmus
This disorder consists of spontaneous horizontal nystagmus, present in the central gaze, which typically reverses direction approximately every 2 minutes.1 During each half cycle, the velocity of slow phases of nystagmus builds up to a maximum, which is clinically evident from prominent quick phases. The nystagmus slow-phase velocity then declines to zero and subsequently reverses the direction; the cycle repeats. Such cyclic oscillation of the slow-phase velocity approximates a sine wave (►Fig. 5).

- An example of slow-phase velocity of periodic alternating nystagmus from two patients. Although average cycle duration of PAN is typically considered 2 minutes, the cycle duration varies amongst patients. Two extreme examples are illustrated here. The slow-phase velocity is plotted in colored symbols along y-axis, while corresponding time is on the x-axis. The trend of slow-phase velocity follows a sinusoidal fit function (gray line). There was a remarkable variability in the cycle duration (18 and 240 seconds) in these two examples of the patients with the same diagnosis (ataxia-telangiectasia) and comparable cerebellar atrophy. This figure was reproduced from Shaikh, et al.40 with permission.
Originally considered a rare and mysterious ocular oscillation, it has provided broad insights into the way that the cerebellum normally governs the temporal properties of the vestibular reflexes. Early on, it was recognized that periodic alternating nystagmus (PAN) occurs in patients with midline cerebellar lesions.37 However, the first insight into its pathogenesis came from the observation that it resembled the nystagmus induced in normal subjects by sustained rotation in darkness at a constant velocity, and the reversal of nystagmus due to an adaptive process, i.e., the role of the velocity storage.16-18 The time course of the initial nystagmus followed by its reversal induced by sustained rotation is similar to that of PAN, suggesting that this nystagmus somehow arises when a vestibular “repair mechanism” goes wrong.37 Further insight into the pathogenesis of PAN came from experimental studies in monkeys. Ablation of the cerebellar nodulus and uvula caused prolonged nystagmus induced by rotation, implying excessive velocity storage (prolonged vestibular memory).17,38 When such monkeys were placed in darkness, they developed PAN; when allowed to view their stationary environment, they visually suppressed this nystagmus.17,38 In human patients with PAN, cerebellar lesions are not only restricted to the nodulus and ventral uvula but also affect other parts of the vestibular cerebellum, such as the flocculus and paraflocculus, which are necessary for normal visual fixation.1 Finally, pharmacological evidence suggests that the nodulus and uvula govern the velocity storage mechanism by exerting inhibitory control on the vestibular commissure via the metabotropic GABAB receptor. Thus, the velocity-storage phenomenon in normal monkeys is suppressed by the GABAB agonist baclofen, and baclofen abolishes PAN in most patients who takes this medicine.18,39
Unusual variants of PAN provided further insight into their pathophysiology. The cycle duration of PAN ranges between 30 seconds and 4 minutes in eight patients with ataxia-tel- angiectasia.40 The two extreme examples are illustrated in ►Fig. 5. In example 1, the cycle duration was 240 seconds, while in example 2 it was approximately 18 seconds. Both patients had ataxia-telangiectasia with comparable amounts of cerebellar atrophy. Then what describes such variability in the cycle length? According to the control systems model of PAN, the cycle duration reflects the dysfunctional “recalibration” through the adaptive mechanism. In these models, the alterations in adaptation gain affects the cycle duration of PAN. Therefore, it is believed that the intrinsic membrane property of the medial vestibular nucleus neurons, a putative site of the velocity storage, shapes the responses to the sensory stimuli, modulating the rate of adaptation, hence the cycle duration of PAN.
Thus, by combining behavioral measurements, experimental lesions, membrane electrophysiology, and pharmacological studies, insights were gained into how adaptive mechanisms, governed by the cerebellum and intrinsic membrane physiology, can function normally, go awry, and be treated with drugs when they go awry. Genetic susceptibility may describe why only “selected” individuals develop disease phenotype and others do not, although the structural lesion or metabolic impairment are the same.
Vertical Nystagmus
The mechanisms for the vertical nystagmus, both downbeat nystagmus (DBN) and upbeat nystagmus (UBN), concern the disturbance of the central pathways conveying vertical vestibulocular reflex.41,42 However, it is most likely that central imbalance of otolithic projections causes UBN,1 because the direction of nystagmus in such patients are often dependent upon head position and modulated by vergence (otolith-ocular responses are strongly modulated by viewing distance).43,44
Here I discuss a common hypothetical mechanism for UBN and DBN and how gravitational force can modulate it. In the simplest terms, there is a “push-pull” mechanism in the pathways regulating vertical eye movements. These forces are comprised of an elastic force due to orbital soft tissue in both upward and downward orientation and an oculomotor integrator for vertical gaze holding. The “push-pull” forces are aligned along the vertical axis in the head-fixed reference frame (blue arrow on caricatures in ►Fig. 6). The “push- pull” forces, in the vertical plane, must be properly “tuned” to account for additional constant (space-fixed) gravitational force. Such tuning could be provided by the “internal estimate of the gravity.” The direction of the internal estimate of the gravity is schematized as the red arrow in ►Fig. 6. In the health, while the subject is oriented upright, the two vectors, the estimate of gravity and push-pull forces (blue and red arrows) are aligned, and gaze would be stable. When the healthy subjects pitch the nose up (as if looking at the sky) in complete darkness, the absence of alternate cues causes a mismatch between the force pulling down and up by cosine of the pitching angle (since internal estimate of the gravity is no longer aligned with the head-fixed vertical axis). Latter would cause downward drifts of the eyes. Conversely, pitching the nose down (as if looking at the ground) would cause upward drifts due to the opposite phenomenon. Such gravity-dependent modulation of the slow-phase velocity is known in healthy subjects (schematic in ►Fig. 6).32 The similar gravity-dependent modulation is seen in patients with DBN or UBN, but the slow-phase velocities are much larger, and depending upon the type of nystagmus there is an offset of the sinusoidal modulation in a specific (upward or downward) direction (►Fig. 6).32 The offset can be described by the “misrepresentation” of the internal estimate of gravity (►Fig. 6). For example, DBN could occur if the internal estimate of gravity is misrepresented as illustrated in ►Fig. 6. In the latter condition, the direction of the gravity estimate shifts farther away as the subject pitches the nose up (►Fig. 6); thus, nystagmus would worsen. Nystagmus would improve when the diseased subject would pitch the nose down because misrepresented internal estimate and push-pull forces are now oriented close to each other. Indeed, DBN often worsens with nose-up orientation and improves in nose-down orientation. The same misrepresentation of the internal estimate of gravity but in the opposite orientation would cause UBN (►Fig. 6). As anticipated, the UBN is often minimal with nose pitched up as compared with pitch down (prone).45,46 Depending upon the amount of misrepresentation (angle between impaired internal estimate of gravity and head vertical axis), beyond certain head orientation, the DBN would be converted to UBN and vice versa, as seen in some cerebellar patients.47

- Schematic diagram illustrating the hypotheses for upbeat nystagmus (UBN) and downbeat nystagmus (DBN). The caricatures on the left side of the diagrams illustrate head orientations: blue arrows depict the direction of the head-fixed push-pull forces (pp), while red arrows illustrate the internal representation of gravity (g). Normally, in darkness, when subjects are upright the push-pull forces align with the internal representation of gravity, but this alignment is disrupted on tilting up or down resulting in slow-phase eye velocity (as illustrated in schematic graph on the right side). Misrepresentation of the internal estimate of gravity results in UBN or DBN.
What are the neural correlates of the internal estimate of gravity? We now know that the firing rates of the canal-otolith convergent neurons at the brainstem vestibular nuclei and rostral fastigial nuclei represent the physical laws of motion and discriminates the gravity from other otolith coded accelerations.48-52 These are the same group of neurons which are thought to comprise vestibular velocity storage and are under inhibitory influence of the cerebellar nodulus.49,50,52 Any nonphysiological change in the firing rate, either due to external disinhibition (e.g., lesion of cerebellar Purkinje neurons) or from alterations in the intrinsic membrane properties (e.g., pharmacological or immune related), can disrupt the “calibration” of the neuronal firing rate to the estimate of gravity; the estimate of gravity would then be skewed.31 Misrepresented internal estimate of gravity would then cause gravity-dependent UBN or DBN. Focal cerebellar lesion affecting cerebellar flocculus can frequently cause DBN, while UBN has been described in focal lesions of brainstem vestibular pathways.1
Funding
The author is supported by the Career Award from The American Academy of Neurology, George C. Cotzias Memorial Fellowship from the American Parkinson's Disease Association, Dystonia Medical Research Foundation Research Grant, and Dystonia Coalition NIH U54 TR001456.
Conflict of Interest
None declared.
References
- Loss of the neural integrator of the oculomotor system from brain stem lesions in monkey. J Neurophysiol. 1987;57(5):1383-1409.
- [CrossRef] [PubMed] [Google Scholar]
- Generation of torsional and vertical eye position signals by the interstitial nucleus of Cajal. Science. 1991;252(5012):1551-1553.
- [CrossRef] [PubMed] [Google Scholar]
- Analysis of the activity of the chains of internuncial neurons. Journal of Neurophysiology. J Neurophysiol. 1938;1(3):207-244.
- [CrossRef] [Google Scholar]
- Nystagmus induced by pharmacological inactivation of the brainstem ocular motor integrator in monkey. Vision Res. 1999;39(25):4286-4295.
- [CrossRef] [PubMed] [Google Scholar]
- The effect of cerebellectomy on the cat’s bestibulo-ocular integrator. Brain Res. 1974;71(2)(3):195-207.
- [CrossRef] [PubMed] [Google Scholar]
- A learning network model of the neural integrator of the oculomotor system. Biol Cybern. 1991;64(6):447-454.
- [CrossRef] [PubMed] [Google Scholar]
- An improved neural-network model for the neural integrator of the oculomotor syste more realistic neuron behavior. Biol Cybern. 1985;53(2):93-108.
- [CrossRef] [PubMed] [Google Scholar]
- In vivo intracellular recording and perturbation of persistent activity in a neural integrator. Nat Neurosci. 2001;4(2):184-193.
- [CrossRef] [PubMed] [Google Scholar]
- Functional dissection of circuitry in a neural integrator. Nat Neurosci. 2007;10(4):494-504.
- [CrossRef] [PubMed] [Google Scholar]
- Spatial gradients and multidimensional dynamics in a neural integrator circuit. Nat Neurosci. 2011;14(9):1150-1159.
- [CrossRef] [PubMed] [Google Scholar]
- Ocular motor abnormalities in hereditary cerebellar ataxia. Brain. 1976;99(2):207-234.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of ablation of flocculus and paraflocculus of eye movements in primate. J Neurophysiol. 1981;46(4):878-899.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebellar control of ocular gaze stability. Ann Neurol. 1980;7(1):37-40.
- [CrossRef] [PubMed] [Google Scholar]
- Velocity storage in the vestibulo-ocular reflex arc (VOR) Exp Brain Res. . 1979;35(2):229-248.
- [CrossRef] [PubMed] [Google Scholar]
- Dynamic modification of the vestibulo-ocular reflex by the nodulus and uvula. Science. 1985;228(4696):199-202.
- [CrossRef] [PubMed] [Google Scholar]
- Baclofen and velocity storage: a model of the effects of the drug on the vestibulo-ocular reflex in the rhesus monkey. J Physiol. 1987;393:703-725.
- [CrossRef] [PubMed] [Google Scholar]
- Effects of baclofen on the angular vestibulo-ocular reflex. Exp Brain Res. 2006;171(2):262-271.
- [CrossRef] [PubMed] [Google Scholar]
- Stimulation of the nodulus and uvula discharges velocity storage in the vestibulo-ocular reflex. Exp Brain Res. 1994;102(1):57-68.
- [CrossRef] [PubMed] [Google Scholar]
- Determinants of spatial and temporal coding by semicircular canal afferents. J Neurophysiol. 2005;93(5):2359-2370.
- [CrossRef] [PubMed] [Google Scholar]
- Role of abducens neurons in vestibuloocular reflex. J Neurophysiol. 1973;36(4):724-738.
- [CrossRef] [PubMed] [Google Scholar]
- Multiple reference frames for motion in the primate cerebellum. J Neurosci. 2004;24(19):4491-4497.
- [CrossRef] [PubMed] [Google Scholar]
- Role of Cerebellum in motion perception and vestibulo-ocular reflex-similarities and disparities. Cerebellum. 2013;12(1):97-107.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebellar infarction presenting isolated vertigo: frequency and vascular topographical patterns. Neurology. 2006;67(7):1178-1183.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebellar infarction—a presentation of vertigo. Laryngoscope. 1980;90(3):505-514.
- [CrossRef] [PubMed] [Google Scholar]
- Motion perception without nystagmus—a novel manifestation of cerebellar stroke. J Stroke Cerebrovasc Dis. 2014;23(5):1148-1156.
- [CrossRef] [PubMed] [Google Scholar]
- Ageotropic central positional nystagmus in nodular infarction. Neurology. 2009;73(14):1163.
- [CrossRef] [PubMed] [Google Scholar]
- Apogeotropic central positional nystagmus as a sole sign of nodular infarction. Neurol Sci. 2012;33(5):1189-1191.
- [CrossRef] [PubMed] [Google Scholar]
- Central paroxysmal positional vertigo: isolated dizziness caused by small cerebellar hemorrhage. Stroke. 2007;38(6):e26-e27. author reply e28
- [CrossRef] [PubMed] [Google Scholar]
- Ataxia telangiectasia: a “disease model” to understand the cerebellar control of vestibular reflexes. J Neurophysiol. 2011;105(6):3034-3041.
- [CrossRef] [PubMed] [Google Scholar]
- Gravity dependence of ocular drift in patients with cerebellar downbeat nystagmus. Ann Neurol. 2002;52(6):712-721.
- [CrossRef] [PubMed] [Google Scholar]
- Familial positional downbeat nystagmus and cerebellar ataxia: clinical and pathologic findings. Ann NY Acad Sci. 2005;1039:540-543.
- [CrossRef] [PubMed] [Google Scholar]
- Cerebellar disease alters the axis of the high-acceleration vestibuloocular reflex. J Neurophysiol. 2005;94(5):3417-3429.
- [CrossRef] [PubMed] [Google Scholar]
- Directional plasticity of the vestibuloocular reflex in the cat. Ann NY Acad Sci. 1981;374:504-512.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic, pathophysiologic, and therapeutic aspects of benign paroxysmal positional vertigo. Otolaryngol Head Neck Surg. 2004;131(4):438-444.
- [CrossRef] [PubMed] [Google Scholar]
- Periodic alternating nystagmus (nystagmus alternans) and excitability of the vestibular system (article in German) Arch Ohren Nasen Kehlkopfheilkd. . 1959;174(3):182-209.
- [CrossRef] [PubMed] [Google Scholar]
- Role of the flocculus and para- flocculus in optokinetic nystagmus and visual-vestibular interactions: effects of lesions. Exp Brain Res. 1983;50(1):9-33.
- [CrossRef] [PubMed] [Google Scholar]
- Treatment of periodic alternating nystagmus. Ann Neurol. 1980;8(6):609-611.
- [CrossRef] [PubMed] [Google Scholar]
- Gaze fixation deficits and their implication in ataxia-telangiectasia. J Neurol Neuro-surg Psychiatry. 2009;80(8):858-864.
- [CrossRef] [PubMed] [Google Scholar]
- Vertical nystagmus: clinical facts and hypotheses. Brain. 2005;128(Pt 6):1237-1246.
- [CrossRef] [PubMed] [Google Scholar]
- Upbeat nystagmus due to a small pontine lesion: evidence for the existence of a crossing ventral tegmental tract. Eur Neurol. 2005;54(4):186-190.
- [CrossRef] [PubMed] [Google Scholar]
- Vestibu- lo-ocular responses to vertical translation in normal human subjects. Exp Brain Res. 2008;185(4):553-562.
- [CrossRef] [PubMed] [Google Scholar]
- A reinterpretation of the purpose of the translational vestibulo-ocular reflex in human subjects. Prog Brain Res. 2008;171:295-302.
- [CrossRef] [PubMed] [Google Scholar]
- Paraneoplastic disorders of eye movements. Ann N Y Acad Sci. 2011;1233:279-284.
- [CrossRef] [PubMed] [Google Scholar]
- Paraneoplastic upbeat nystagmus. Neurology. 2011;77(7):691-693.
- [CrossRef] [PubMed] [Google Scholar]
- Effect of 3,4-diaminopyridine on the gravity dependence of ocular drift in downbeat nystagmus. Neurology. 2004;63(4):752-753.
- [CrossRef] [PubMed] [Google Scholar]
- Neurons compute internal models of the physical laws of motion. Nature. 2004;430(6999):560-564.
- [CrossRef] [PubMed] [Google Scholar]
- Resolution of sensory ambiguities for gaze stabilization requires a second neural integrator. J Neurosci. 2003;23(28):9265-9275.
- [CrossRef] [PubMed] [Google Scholar]
- Sensory vestibular contributions to constructing internal models of self-motion. J Neural Eng. 2005;2(3):S164-S179.
- [CrossRef] [PubMed] [Google Scholar]
- Sensory convergence solves a motion ambiguity problem. Curr Biol. 2005;15(18):1657-1662.
- [CrossRef] [PubMed] [Google Scholar]
- Purkinje cells in posterior cerebellar vermis encode motion in an inertial reference frame. Neuron. 2007;54(6):973-985.
- [CrossRef] [PubMed] [Google Scholar]
- Cervical dystonia: a neural integrator disorder. Brain. 2016;139(Pt 10):2590-2599.
- [CrossRef] [PubMed] [Google Scholar]




